|
Abstract
Extraskeletal bone tumors are rare and high grade tumors
including osteosarcoma, chondrosarcoma and Ewings sarcoma of
the soft tissues and its variants. A retrospective study of 25
cases of these tumors was made in our institution in the 1983 -
2000 period. The study of 25 cases revealed that these tumors
affect adults (median age 51.2, range 17-73 years). The most
common tumor locations were the thigh (12 cases, 48%) and the
arm-elbow (6 cases, 24%). As a classification for diagnostic
type of tumors: 14 were chondrosarcomas (56%), 8 were
osteosarcomas (32%) and 3 were Ewing sarcomas (12%). The median
follow-up was 47.07 months with a range from 24 months to 156
months. All the cases were treated with preop. chemotherapy and
postoperative radiotherapy (with the exception of the myxoid
chondrosarcoma). Globally, the preoperative duration of symptoms
ranged from 2 weeks to 6 years (median 6 months). Local
recurrences after wide and compartimental margin surgery
developed in 7 cases (12%), three cases of chondrosarcoma and
four cases of extraskeletal osteosarcoma; and distant metastases
developed in seven cases (6 osteosarcomas and 1 Ewings
sarcoma). The two year overall survival rates were:
extraeskeletal chondrosarcoma 50%, osteosarcoma 36.5% and Ewing
sarcoma 66.6%. The two year disease-free survival was: 42.8%
chondrosarcoma, 25% osteosarcoma and 33.3% Ewing sarcoma. The
interest of this series is the fact that tumors are high grade
and cure may be achieved by wide or compartimental local
excision of the tumor at an early stage of the disease (combined
with radiation and chemotherapy).
J.Orthopaedics 2006;3(4)e7
Introduction:
Extraskeletal primary bone sarcomas are rare
and high-grade tumors that include osteosarcoma (OS),
chondrosarcoma (CHO) and Ewing sarcoma (EW) of the soft tissues
and its variants.
Osteosarcoma of the soft tissues is a
malignant mesenchymal tumor whose cells produce osteoid
substance. Unlike skeletal osteosarcoma it is observed during
adult and advanced age and particularly in women. The sites most
involved are the thighs, the muscles of the pelvic girdle and
the shoulder. Radiographically, there may be areas or nodules of
faced radiopacity, due to tumoral osteogenesis. Microscopically,
skeletal osteosarcoma (OS) is prevalently osteoblastic, but
chondroblastic and/or fibroblastic areas may be present.
Differential diagnosis must include osteproductive benign
lesions (i.e. myossitis ossificans) and malignant lesions (i.e.
sarcomas with osteogenesis of a metaplastic nature). The
prognosis is very severe, as pulmonary metastasis frequently
occurs. The treatment is the same as for skeletal OS:
preoperative chemotherapy, wide or radical surgery, and
maintenance chemotherapy.
Chondrosarcoma of the soft tissues is very
rare. Instead, almost all the cases of CHO are constituted by
myxoid forms and by mesenchymal chondrosarcoma.
Extraskeletal myxoid chondrosarcoma (named
chordoid sarcoma) is a rare tumor that affects men more than
women and it is almost exclusively observed during adult and
advanced age. It is specially observed deeper in the lower limb.
The radiographic picture is completely unspecific, locking
images of calcification or ossification. Histologically, it has
a lobular structure, more or less cellular; nonetheless this
differentiation hardly ever achieves the stage of
well-differentiated hyaline cartilage. Differential diagnosis
must include myxoid liposarcoma, myxoma and chordoma. The
prognosis is the same as that for skeletal chondrosarcoma, but
it has a considerable tendency to recur locally and it is
capable of metastasizing. The most suitable type of treatment
appears to be wide surgical excision.
Mesenchymal chondrosarcoma is rare in the
soft tissues, even more rare than in the skeleton. There is no
predilection for sex, and unlike myxoid chondrosarcoma it is
observed during young and adult age (15-40 years) and deeper in
the lower limb and neck. Clinically, again unlike myxoid
chondrosarcoma, it has a rather rapid growth. Radiographic
appearance is often sprayed by calcifications, with angiography
the tumor is injected intensely, in virtue of its rather rich
and dilated capillary circulation. Histologically, the pattern
is intense vascularization and dilated sinusoidal with
balloon-oval cells by the presence of foci of cartilaginous
differentiation. Differential diagnosis above all involves
hemangioperycitoma. Prognosis is very severe (high malignancy).
Its growth is rapid and its tendency to metastasize is also
high. The treatment includes surgery, chemotherapy and
radiation.
Synovial chondrosarcoma are exceptional cases
in which a synovial chondromatosis causes a chondrosarcoma, or
in which a chondrosarcoma originates in the joint. It is a soft
and compact cartilaginous tissue filling the joint cavity,
eroding the capsule, invading the soft tissues, digging into and
infiltrating the joint bones. It is difficult to establish
whether it was truly a chondrosarcoma or an aggressive and
tumor-like synovial chondromatosis.
Ewing sarcoma of the soft tissues remains a
rare tumor, it shows predilection for the male sex, and for
those aged between 15 and 45 years. Histologically, the aspect
is the same as that of Ewing sarcoma of the bone: uniform fields
of small and round cells. Differential diagnosis must include
neuroblastoma,embryonal or alveolar rhabdomyosarcoma, and for
patients aged over 30-50 years malignant lymphoma and metastasis
of small cell carcinoma. Prognosis and treatment seem to be very
similar to what is indicated for Ewing sarcoma of the skeleton.
Method and materials
Retrospective studies of 25 cases of this
heterogenic group of tumors were made in our Hospital in the
1983 - 2000 period (Table I). The study of 25 cases revealed
that these tumors affect adults (median age 51.2, range 17-73
years). The most common tumor locations were the thigh (12
cases, 48%) and the arm-elbow (6 cases, 24%). Three tumors were
superficial (hand and foot locations, 12%), 3 pelvic locations
(12%) and 1 perone location (4%).
As a classification for diagnostic type of
tumors: 14 were chondrosarcomas(56%), 8 were osteosarcomas (32%)
and 3 were Ewing sarcomas (12%) (Table III) and staging with the
AJCC classification (Table II and III).
Fourteen extraskeletal chondrosarcomas are
distributed in 6 myxoid chondrosarcomas: 5 male and 1 female
patients, median age 58.3 years (ratio 47-70 years) with a
median follow-up of 49.5 months (ratio 24-95 months), 5 thigh
and 1 arm locations; 5 mesenchymal chondrosarcomas: 4
females and 1 male patient, median age 33.6 years (ratio 17-56
years) with a median follow-up of 30.2 months (ratio 26-36
months), 3 thighs, 1 arm and 1 foot locations and 3 synovial
chondrosarcomas: 2 male and 1 female patients, median age 47.6
years (ratio 43-55 years) with a median follow-up of 30.6 months
(ratio 28-36 months), 2 knee-thigh and 1 hip-thigh
locations.
The 6 cases of
myxoid chondrosarcoma were treated initially with wide and
compartimental surgery (three wide surgeries and three
compartimental surgeries without adjuvant and coadjuvant
treatment). The 5 cases of mesenchymal chondrosarcoma were
treated initially with wide and compartimental surgery (four
compartimental surgeries and one wide surgery) accompanied with
coadjuvant chemotherapy and radiation. The 3 cases of synovial
chondrosarcoma were treated initially with wide surgery
associated in this kind of tumor, in one case with coadjuvant
chemotherapy and in another case with postoperative radiation;
in one case supracondylar amputation is performed and in another
case hemipelvectomy is preformed for tumor local persistence.
|
STAGE |
GRADE |
TUMOR (T) |
LIMPHATIC
NODES(N) |
METASTASIS
(M) |
|
IA |
Low |
T1a-1b |
N0 |
M0 |
|
IB |
Low |
T2a-2b |
N0 |
M0 |
|
IIA |
High |
T1a-1b |
N0 |
M0 |
|
IIB |
High |
T2a |
N0 |
M0 |
|
III
|
High |
T2b |
N0 |
M0 |
|
IV |
Low and High |
T1-2
T1-2 |
N1
N0-1 |
M0
M1 |
T1a
Superficial
T1b Deep
T2 > 5 cm
T2a Superficial
T2b Deep
N1 Regional limph nodes
G 1-2 Low grade
G 3-4 High grade.
Table II.
Clasificación UICC/AJCC 2002 (VI edition).
Eight
extraskeletal osteosarcomas were revised: 6 male and 2 female
patients, median age 58 years (ratio 28-73 years) with a median
follow-up of 73.12 months (ratio 24-156 months), 3 arm-elbow, 2
pelvic, 1 arm-shoulder, 1 thigh and 1 perone locations. All of
the cases of osteosarcomas were treated initially with
preoperative or neoadjuvant chemotherapy after surgery (1 case
of initial leg supracondylar amputation, 4 cases of initial wide
surgery and 1 initial compartimental surgery were performed) and
postoperative or coadjuvant chemotherapy and postoperative
radiotherapy (except the case of initially amputation
performed). The two cases of pelvic locations were treated with
chemotherapy and radiation only (surgery is not possible in
these particular cases).
Three
extraskeletal Ewing sarcomas were studied: 3 female patients,
median age 51.6 years (ratio 46-62 years) with a median
follow-up of 29 months (ratio 24-33 months), 2 foot and 1 pelvic
locations. All the cases were treated initially with neoadjuvant
chemotherapy after surgery (2 cases of initial infracondylar
amputations in foot locations and abstention of surgery in
pelvic location) and coadjuvant chemotherapy and local
radiation.
Globally, the
preoperative duration of symptoms ranged from 2 weeks to 6 years
(median 6 months). The median follow-up was 47.07 months with a
range from 24 months to 156 months.
One case of
osteosarcoma presented a history of previous radiation and 2
cases of osteosarcoma presented a history of prior trauma.
|
Name
Diagnosis |
|
AJCC
|
|
AGS |
Myx-CHO |
II B |
|
JSS
|
Mes-CHO |
III |
|
JSA |
Myx-CHO |
II C |
|
ERM |
Mes-CHO |
III |
|
AML |
Myx-CHO |
III |
|
MTT |
Mes-CHO |
III |
|
PFP |
Myx-CHO |
II C |
|
JGB |
Myx-CHO |
II B |
|
ABN |
Mes-CHO |
III |
|
LSV |
Mes-CHO |
III |
|
AMO |
Myx-CHO |
II B |
|
MRP
|
Synovial-CHO |
III |
|
ROS |
Synovial-CHO |
III |
|
JMC |
OS |
III |
|
JVB |
OS |
II C |
|
FSM |
OS |
III |
|
JNM |
OS |
III |
|
EMG |
OS |
IV B |
|
AMF |
OS |
III |
|
EMG |
OS |
IV B |
|
AMF |
OS |
II C |
|
JOA |
EW |
III |
|
PCL |
EW |
IV A |
|
ASS |
EW |
III |
|
MAC |
EW |
III |
Table III. Diagnostic
type of tumors and AJCC classification.
Results
Treatment complications
In the 25 cases presented, the following treatment complications
were observed: 2 superficial infections, 2 toxicity
chemotherapy, 2 ulcerated tumors with recurrence and 1
thromboembolism.
Local recurrence
Local recurrences after wide and compartimental margin
surgery developed in 7 cases (12%). Three cases of
chondrosarcoma and four cases of extraskeletal osteosarcoma.
In the cases of myxoid, mesenchymal and
synovial chondrosarcoma, eleven cases were alive with no
evidence of recurrence (78.5%).Three cases of recurrences: one
synovial chondrosarcoma of the knee one year after wide
resection, the recurrence was treated with compartimental
surgery and it had a new recurrence two years later the first
wide surgery and its was treated with a above-the-knee
amputation associated with postoperative chemotherapy. This
patient was alive with one or more recurrences and two cases of
mesenchymal chondrosarcomas treated initially with wide surgery
and ampliation of surgery at compartimental where the
recurrence was present at two and one year (one arm location and
one thigh location).
In the cases of extraskeletal osteosarcomas,
four cases were alive with no evidence of recurrence (50%), and
another four cases of recidivated extraskeletal osteosarcomas
were treated; two cases of extraskeletal osteosarcoma in the
arm-elbow developed a recurrence one year and two years later
respectively after wide resection and they were treated with an
above-the-elbow amputation and compartimental surgery
respectively associated with postoperative chemotherapy
(alive with lung metastasis). Another case presented recurrence
in arm-shoulder location two years after wide resection, and it
was treated with compartimental surgery (alive without
metastasis). One case of recurrence of extraskeletal
osteosarcoma of the thigh, one case developed a recurrence 18
months after wide resection and it was treated with a
suprecondylar amputation associated with postoperative
chemotherapy (alive without metastasis).In the cases of
extraskeletal Ewing sarcomas, none were recidivated.
Metastasic disease-Exitus
None of the cases of extraskeletal
chondrosarcomas and its variants were metastasized.
In cases of extraskeletal osteosarcoma, five cases (62.5%)
presented lung metastasis (two cases at diagnosis time) and
three cases (37.5%) died after 134 months (peroneal location),
80 months and 29 months (pelvic location), respectively. The two
cases with metastasic lung disease were arm-elbow locations
treated with wide surgery initially and subsequent amputation
above-the-elbow in one particular case after recurrence. The
other case was treated with wide resection initially. These
cases were alive with metastasic disease (25%) and achieved
local control after 156 months and 89 months respectively.
In extraskeletal Ewing sarcoma cases, one case (33%) of foot
location developed regional lymph nodes metastasis and it is
alive after 14 months at diagnosis. One pelvic case died after
33 months and another foot location case was alive without
metastasis disease after 14 months.
Survival rates for type of tumors
The two year overall survival rates
were: extraeskeletal chondrosarcoma 50%, osteosarcoma 36.5% and
Ewing sarcoma 66.6%. The two year disease-free survival were:
42.8% chondrosarcoma, 25% osteosarcoma and 33.3% Ewing sarcoma.
Statistical analysis
This retrospective study is based in a short series of patients
and a rare and heterogenic group of tumors. The statistical
analysis included the study of several variables: tumor
location(Table IV), type of tumor (Table V), staging system
(Table VI) and surgical treatment (Table VII) with one
curve of global overall survivor (Table VIII). This study is
presented following a Kaplan-Meier system of survivorship
curves.
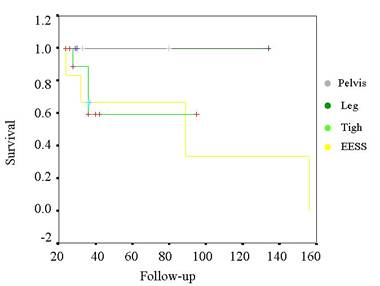
Table IV.
Kaplan-Meier curve for survival of the tumors related by
surgery.
The statistical significance only was
demonstrated in the case of surgical treatment. None of the rest
of variables included in this study (diagnostic tumor, staging
system, and tumor location) are significative statistically.
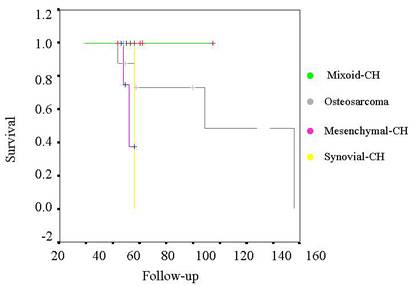
Tabla V. Kaplan-Meier curve for survival
of the tumors related by histology.
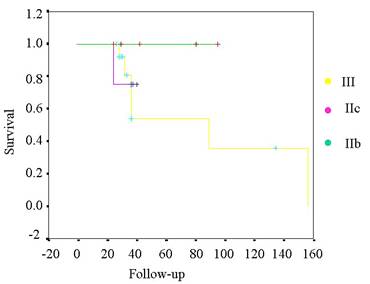
Tabla VI. Kaplan-Meier curve for survival
of the tumors related by staging.
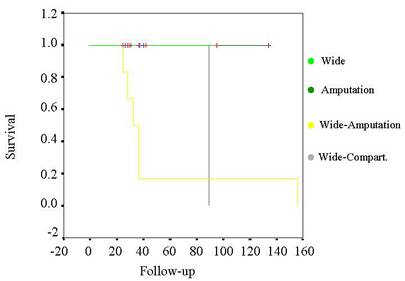
Table
VII. Kaplan-Meier
curve for survival of the tumors related by surgery.
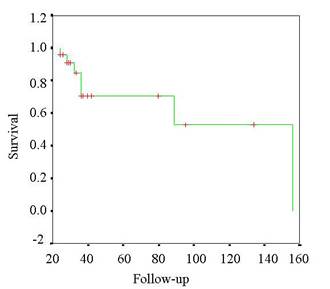
Table VIII.
Kaplan-Meier global overall survivor.
Discussion :
This group of tumors rarely occur and they are
included in the XII group of the W.H.O. classification
(WHO book)
(extraskekeletal bone tumors) in
the cases of osteosarcomas and chondrosarcomas and the group of
lesions of uncertain origin in the case of Ewing sarcoma-PNET
group.
As for osteosarcoma patients, there is a poor type of tumor
prognosis due to the aggressive biological behavior of this kind
of tumors (high tendency to local recurrence and general
dissemination). The staging of these tumors is grade II-C or
high (IV) in the American Joint Committee System Staging
(variation 1997)(
Sugarbaker book,5) : >5 cm tumors, superficial or
deep, with or without lymph and lung metastases. In
previous studies of extraskeletal osteosarcoma
(1,2) the
thigh was the most common tumor location; in our series, the
most common location is the arm-elbow and pelvic tumors (worse
prognosis for a location that prevents surgical treatment). The
extraskeletal osteosarcoma was described in more locations: hand(6),urinary
bladder, prostate, kidney, breast, lung, tiroid,
retroperitoneum
(3), frontal region(4) and
cardiac intramuscular(5) presentations.
Etiology of the cases is controversial, several theories were
performed: association with Li-Fraumeni Syndrome(7) and
myositis ossificans(8) ,
prior trauma(9) ,
radiation-induced
(10) , thorotrast-induced
(11)
and heterotopic ossification after an electrical burn
(12).
Microscopically tumors contain varying amounts of
neoplasic osteoid of bone (Figure 1), sometimes together with
islands of malignant-appearing cartilage
(13).
Figure 1. Histological pattern of extraskeletal
osteosarcoma.
Hematoxyline-
eosine
x 400.
In radiological studies extraskeletal osteosarcoma presented
nodules of faced
radiopacity ,(tumoral osteogenesis)
(14),
central ossification and inverse zone
phenomenon (Figure 2).
Figure 2. Extraskeletal osteosarcoma of
the arm. Radiological appearence.
Bone scan
reveals uptaking in the lesion
(15) CT
scan revealed mineralized soft tissues, and MRI presentation
shows hipointense images in T1-weigthed spin echo and
hiperintense in T2-weigthed spin echo and STIR sequences.
Telangiectatic variant are least common histological variety in
this group (16).
As for the diagnosis of these, it was made by needle biopsy
(tru-cut technique in all the cases), but with appropriate
clinicoradiologic correlation extraskeletal osteosarcoma may not
be recognized easily by FNAB
(17),
unlike skeletal osteosarcoma. Like the osteosarcoma of bone,
extraskeletal osteosarcoma showed a striking variation in
histological appearance and focally resembled malignant fibrous
histiocytoma (1),
fibrosarcoma, soft tissue aneurismal bone cyst
(16 ) and
malignant schwannoma. The prevailing sites of metastases were
the lung, the regional lymph nodes and bone (none in our series
of patients). The treatment included in all the cases
neoadjuvant chemotherapy, surgery (wide, compartimental or
radical) and coadjuvant chemotherapy with local postoperative
radiation. Extraskeletal osteosarcoma is a high grade
malignant tumor associated with a 5-year survival rate of 37% of
the cases. Local recurrences and distant metastasis are common
and usually occur by 3 years after excision in Mayo Clinic
series
(18). In
comparison, in our series the 2-year survival rate is 36.5 % and
the 2-year free of disease survival rate is 25%. Additional
larger series will be required before drawing definite
conclusions.
Chondrosarcoma
of the soft tissues is a rare tumor. The near totality of CHO is
instead constituted by myxoid forms, mesenchymal chondrosarcoma
and by synovial sarcoma.
Extraskeletal
myxoid chondrosarcoma is an uncommon neoplasm, according for
less than 2% of all soft tissue sarcomas. It affects adult males
with an age in the fifth decade at the time of diagnosis. The
tumor usually arises in the deep soft tissues, especially in the
lower extremities (Figure 3)(19).
Figure 3. Extraskeletal myxoid chondrosarcoma. Thigh
location. MRI T1-weighted image.Figure 4. Histological
pattern of extraskeletal myxoid chondrosarcoma.
Hematosyline
-eosine x 200.
It is a rare low-grade soft tissue sarcoma (staging of these
tumor varies in II-B, II-C or III in the AJCC system), with
locally aggressive and metastasizing potential
(20) . It is specially observed deeper in the
lower limbs. In myxoid CHO, the cells that resemble epithelial,
can very closely mimic some malignant breast tumors in thoracic
locations (21).
A diagnosis of extraskeletal myxoid chondrosarcoma was rendered
based on histological findings
(22). Differential diagnosis included other myxoid
neoplasm such as bony myxoid chondrosarcoma, myxoid liposarcoma,
chordoma and parachordoma
(22).
Differential diagnosis
with extaskeletal chondroma: asymptomatic and harmless
clinical course, the lack of connection between the tumor and
the underlying bone, the slow tumor development, the absence of
a sex predominance and the characteristic tumor histological
picture .Typically well-circumscribed, extraskeletal myxoid
chondrosarcomas are commonly encapsulated by a rim of fibrous
tissue. The abundant myxoid matrix gives the cut surface a
gelatinous appearance. The degree of cellularity is variable;
less well-differentiated, highly cellular tumors generally have
less extracellular matrix and behave more aggressively (Figure
4) (22)
Pulmonary metastases are not unusual in this tumor, only two
patients have been reported with multiple bone metastases
(23)
In our series no adjuvant therapy is necessary, none of the
cases were disseminated. Resistance to standard soft tissue
sarcoma chemotherapy has been demonstrated
(24) .
Myxoid CHO are a better prognosis respect to synovial CHO and
mesenchymal CHO
(25).
Figure 5. MRI of
mesenchimal chondrosarcoma
of the thigh.
Figure 6.
Synovial chondromatosis of the hip.
MRI T1-weighted image. Mesenchymal
chondrosarcoma is rare in the soft tissues (making up less than
2% of all chondrosarcomas)
(26), even
more rare than in the skeleton, There is no predilection for
sex, and unlike myxoid chondrosarcoma it is observed during
young and adult age (15-40 years) and deeper in the lower limb
(Figure 5) and neck.
The
extraskeletal mesenchymal chondrosarcoma was described in more
rare locations: jaw
(28) ,
intraspinal (29),
cauda equina (30),
pleura (31),
labium majus (32),
orbit (33),
heart (34),
intracranial (35),
femoral vein (36) and
vagus nerve (37).
Clinically, again unlike myxoid chondrosarcoma, it has a rather
rapid growth. Radiographic appearance is often sprayed by
calcifications, with angiography the tumor is injected
intensely, in virtue of is rather rich and dilated capillary
circulation. Histologically pattern is intense vascularization
and dilated sinusoidal with balloon-oval cells by the presence
of foci of cartilaginous differentiation
(38),
typically characterized by tumor compartmentation
(undifferentiated tumor cells in the small-cell areas were
negative for vimentin and cytoprotein S-100, whereas other tumor
cells expressed collagen type II-A and vimentin indicating a
chondroprogenitor cellular phenotype in these small areas)(39) ,
citogenetic studies of mesenchymal chondrosarcoma are few and to
date, no specific or recurrent aberrations have been found
(40,41, 42,43)
. Differential diagnosis above all involves
hemangioperycitoma. Prognosis is very severe (high malignancy)
(44). Its
growth is rapid and its tendency to metastasize is also high
(lung, bone and skin metastases
(44) ). The treatment includes surgery, chemotherapy and
radiation.
Synovial chondrosarcoma are exceptional cases in which a
synovial chondromatosis causes a chondrosarcoma
(45, 46), or in which a chondrosarcoma originates in the
joint. It is a soft and compact cartilaginous tissue filling the
joint cavity, eroding the capsule, invading the soft tissues,
digging into and infiltrating the joint bones (Figure 6).
Malignant transformation to chondrosarcoma shows no specific MR
features to distinguish these cases with malignant change of
primary synovial chondromatosis alone
(45).
Synovial chondrosarcoma is considered very rare and it is not
always clear whether the sarcoma develops by malignant
transformation of synovial chondromatosis or whether it arises
de novo. Differentiation of the two conditions based on clinical
and radiographic features is not possible and it can be
difficult (46)
based on histological criteria. The indispensable
criteria to diagnose malignant transformation are: 1)
histological diagnosis of synovial chondromatosis established
before diagnosis of chondrosarcoma, 2) histological diagnosis of
chondrosarcoma on the same anatomic site as the synovial
chondromatosis, 3) diagnosis of chondrosarcoma and synovial
chondromatosis on the same resection specimen(47).
The mo st important histological features were loss of the
clustering growth pattern typical of synovial chondromatosis,
myxoid change in the matrix, areas of necrosis, and splinting at
the periphery of chondroid lobules (Figure 7)
(48) . It
is difficult to establish whether it was truly a chondrosarcoma
or an aggressive and tumor like synovial chondromatosis. This
condition was described in the hip joint
(49), knee
joint (50)
, radiocarpal joint
(51), ankle
joint (52) and
metacarpophalangeal joint
(53).
Extraskeletal
Ewing sarcoma (EES) is a rare soft tissue tumor that is
morphologically indistinguishable from the more common Ewing
sarcoma of bone (Figure 8) .
Figure 7.
Histological appearence of synovial
chondrosarcoma.
Hematoxyline-eosine x400.Figure
8. Histological
pattern of extraskeletal
Ewing sarcoma. Hematoxyline-eosine x400.
It must be
distinguished from other small, blue round cell tumors (SBRCTs).
The most frequent sites of occurrence are the chest wall, lower
extremities, and paravertebral region. Less frequently, the
tumor occurs in the pelvis and hip region, the retroperitoneum,
and the upper extremities. It is usually found in young adults
(younger than 30 years) and has a slight predominance in male
patients (54).
Ewing sarcoma is the second most common primary osseous
malignancy in childhood and adolescence, and most of the
success in survival with multimodality treatment has been in
that age group. Survival rates in patients with childhood
nonmetastatic ES/PNET have improved from 10% in 1967(55) to
50% in 2000(57,58) The
improvement in survival is primarily associated with the
combination of surgery and chemotherapy. Little has been
pubfished about the outcome of adults with extraosseous (soft
tissue) ES/PNET.
Four studies to
date, regarding both skeletal and extraskeletal adult
ES/PNET(Figure 9), have been published evaluating survival and
predictors of survival. Two studies(59,60) have
demonstrated age as a poor predictor of outcome, and 2 studies(61,62) have
not seen age as an adverse risk factor. Our extraskeletal Ewing
series shows a higher median age (51.6 years) and the courious
location (2 foot cases and 1 pelvic case).
Figure 9.
Histological appearence of PNET sarcoma.
Hamatoxyline-eosine x 200.
Conclusion
Extraskeletal bone tumors are tumors
predominantly high-grade tumors (except the facto f myxoid
chondrosarcoma).The treatment includes preoperative
chemotherapy, surgery and postoperative chemotherapy and
radiotherapy in the cases of OS, EW and mesenchymal CHO. CHO was
treated with radical, wide and compartimental margins surgery.
The prevailing
sites of metastasis were the lung and the regional lymph nodes.
No bone metastasis was registered.
Cure may be
achieved by wide or compartimental local excision of the tumor
at an early stage of the disease (combined with radiation and
chemoterapy in the cases of OS, Ewand myxoid CHO).
Myxoid CHO have
better prognosis than synovial CHO and mesenchymal CHO.
In myxoid CHO,
the cells that resemble epithelial cells, can very closely mimic
some malignant breast tumors in thoracic locations.
Differential
diagnosis with extaskeletal chondroma: asymptomatic and harmless
clinical course, the lack of connection between the tumor and
the underlying bone, the slow tumor development, the absence of
sex predominance and the characteristic tumor histological
picture.
Pulmonary
metastasis is not usual in extraskeletal myxoid chondrosarcoma,
only two patients have been reported with multiple bone
metastasis.
Biopsy was made
in all cases for punction biopsy with the tru-cut technique.
Extraskeletal
osteosarcoma is the poor type of tumor prognosis (three cases of
exitus, three cases of local recurrence and five cases of lung
metastases).
Additional larger series will be required
before drawing definite conclusions
Reference :
-
Jensen ML, Schumaker B, Jensen OM, Steen JM.
Extraskeletal osteosarcomas: a clinicopathologic study of 25
cases. Am J Surg Pathol 1998; 22:588-94.
-
Chung EB, Enzinger FM. Extraskeletal
osteosarcoma. Cancer 1987; 60: 1132-42.
-
Van Rijswijk CSP, Lieng JGSTA, Kroon HM,
Hogendoorn PCW. Retroperitoneal extraskeletal osteosarcoma. J
Clin Pathology 2001; 54:77-78.
-
Lima MA, Rivas LG., Grecco MA, Drumond JM.
Extraskeletal primary osteosarcoma of the frontal region. Rev
Assoc Med Bras 1998; 44: 43-6.
-
Sunil KS, Ammash MN. MD; Edwards WD. MD; Breen
JF. MD; Edmonson J MD. Calcified Left Ventricular Mass: Unusual
Clinical, Echocardiographic, and Computed Tomographic Findings
of Primary Cardiac Osteosarcoma. Mayo Clinic Proceedings 2000;
75: 743-747.
-
Cook PA, Murphy MS, Innis PC, Yu JS.
Extraskeletal Osteosarcoma of the Hand. A Case Report. J Bone
Joint Surg Am 1998; 80A: 725-729.
-
Frenourg T, Abel A; Bonaiti-Pellie C, Brugieres
L, Berthet P, Bressac -de Paillerets B, Chevrier A, Chompret A,
Cohen-Haguenauer O, Delattre O, Feingold J, Feunteun J, Frappaz
D, Fricker JP, Gesta P, Jonveaux P, Kalifa C, Lasset C, Leheup
B, Limacher JM, Longy M, Nogues C, Oppenheim D, Sommelet D,
Soubrier F Stoll C, Stoppa-Lyonnet D, Tristant H. Li-Fraumeni
syndrome: update, new data and guidelines for clinical
management. Bulletin du Cancer 2001; 88:581-7.
-
Konishi E, Kusuzaki K, Murata H, Tsuchihashi Y,
Beabout JW, Unni KK. Extraskeletal osteosarcoma arising in
myositis ossificans. Skeletal Radiology 2001;. 30:39-43.
-
Puzas JE, Miller MD, Rosier RN. Pathologic Bone
Formation. Clin Orthop 1998; 245: 269-279.
-
Alpert LI, Abaci IF, Werthamer S.
Radiation-induced extraskeletal osteosarcoma. Cancer 1973;
31:1359-63..
-
Hasson J, Hartman KS, Milikov E, Mittelman JA.
Thorotrast-induced extraskeletal osteosarcoma of the cervical
region. Report of a case. Cancer 1975; 36:1827-33.
-
Aboulafia AJ, Brooks F, Piratzky J, Weiss S.
Osteosarcoma Arising from Heterotopic Ossification After an
Electrical Burn: A Case Report. J Bone Joint Surg 1999; 81-A:
564-570.
-
Kransdorf MJ, Meis JM. From the archives of the
AFIP. Extraskeletal osseous and cartilaginous tumors of the
extremities. Radiographics 1993; 13: 853-84.C
-
ampanacci M. Bone and Soft Tissue Tumors.
Springer-Verlag Wien New York, pp. 1039-1042.
-
Arend SM, Pawels EK, Ardnt JW, Meinders AE.
Extraskeletal localization of the 99m Tc-labeled bone-seeking
tracers in bone scintigraphy. Neth J Med 1994; 45: 177-91.
-
Nielsen GP, Fletcher CD, Smith MA, Rybak L,
Rosemberg AE. Soft tissue aneurysmal bone cyst: a
clinicopathologic study of five cases. Am J Surg Pathol 2002;
26:64-9.
-
Nicol KK, Ward WG, Savage PD, Kilpatrick SE.
Fine-needle aspiration biopsy of skeletal versus extraskeletal
osteosarcoma. Cancer 1998; 8: 176-85.
-
Lee JS, Fetsch JF, Wasdhal DA, Lee BP, Pritchard
DJ, Nascimento AG. A review of 40 patients with extraskeletal
osteosarcoma. Cancer 1995; 76: 2253-9.
-
Gebhardt MC, Parekh SG, Rosemberg AE, Rosenthal
DI. Extraskeletal myxoid chondrosarcoma of the knee. Skeletal
Radiol 2000; 29: 302-3.
-
Goh YW, Spagnolo DV, Platten M., Caterina P,
Fisher C, Oliveira AM, Nascimento AG. Extraskeletal myxoid
chondrosarcoma: a ligth microscopic, inmunohistochemical,
ultrastructural and inmunostructural study indicating
neuroendocrine criteria. Histopathology 2001; 39: 514-24.
-
Rao L, Kudva R, Rao RV, Kumar B. Extraskeletal
myxoid chondrosarcoma of the chest wall masquerading as a breast
tumor. A case report. Acta Cytol 2002; 46: 417-21.
-
Partham DM. Pathologic case of the month.
Diagnosis and Discussion: Extraskeletal Myxoid Chondrosarcoma
.Archiv Pediat Adolescent Med 1999; 153.
-
Takeda A, Tsuchiya H, Mori Y, Nonomura A, Tomita
K.Extraskeletal myxoid chondrosarcoma with multiple skeletal
metastases. J Orthop Sci 2000; 5: 171-4.
-
Patel SR, Burgess MA, Papadopoulous NE.
Extraskeletal myxoid chondrosarcoma: long-term experience with
chemotherapy. Am J Clin Onco 1995; 18: 161-163.
-
Saleh G., Evans HL, Ro JY, Ayala AG.
Extraskeletal myxoid chondrosarcoma: a clinicopathologic study
of ten patients with long-term follow-up. Cancer 1992; 70:
2827-2830.
-
Theodorou DJ, Theodorou SJ, Xenaquis T, Demou S,
Agnantis N, Soucacos PN. Mesenchymal chondrosarcoma of soft
tissues of the calf. Am J Othop 2001; 30: 329-32.
-
Suryanarayana KV, Balkrishnan R, Rao L, Rahim
TA. Parapharyngeal space mesenchymal chondrosarcoma in
childhood. Intern J Ped Otorhinolaryngology 1999; 50: 69-72.
-
Takahashi K, Sato K, Kanazawa H, Wang XL, Kimura
T. Mesenchymal chondrosarcoma of the jaw. Rport of a case and
review of 41 cases in the literature. Head & Neck 1993; 15:
459-64.
-
Ranjan A, Chacko G, Joseph T, Chandi SM.
Intraspinal mesenchymal chondrosarcoma. Case report. J Neurosurg
1994; 80: 928-30.
-
Rushing EJ, Mena H, Smirniopoulos JG.
Mesenchymal chondrosarcoma of the cauda equina.Clin Neuropathol
1995; 14:150-3.
-
Luppi G, Cesinaro AM, Zoboli A, Morandi U,
Piccinini L. Mesenchymal chondrosarcoma of the pleura. Europ
Resp J 1996; 9: 840-3.
-
Lin J, Yip KM, Maffulli N, Chow LT.
Extraskeletal mesenchymal chondrosarcoma of the labium majus.
Gynecol Oncol 1996; 60:492-3.
-
Khouja N, Ben Amor S, Jemel H, Kchir N, Boussen
H, Khaldi M. Mesenchymal extraskeletal chondrosarcoma of
the orbit. Report of a case and review of the literature. Surg
Neurol 1999; 52:50-3.
-
Nesi G, Pedemonte E, Gori F. Extraskeletal
mesenchymal chondrosarcoma involving the heart: report of a
case. Italian Heart J 2000; 1: 435-7.
-
Bingaman KD, Alleyne CH Jr, Olson JJ.
Intracranial extraskeletal mesenchymal chondrosarcoma: a case
report. Neurosurgery 2000; 46:207-11.
-
Kin GE. Kim do K, Park IJ, Ro JY. Mesenchymal
chondrosarcoma originating from the femoral vein. J Vasc Surg
2003; 37: 202-5.
-
Jamal W, Rhys Evans P, Sheppard MN. Mesenchymal
chondrosarcoma of the vagus nerve. J Laryngology & Otology 2002;
116: 644-6.
-
Schorle CM, Unni KK, Aigner T. Neoplastic
chondroneogenesis as a characteristic of mesenchymal
chondrosarcomas. Orthopade 2002; 31:544-50.
-
Naumann S, Krallman PA, Unni KK, Fidler JR,
Bridge JA. Translocation der (13;21)(q10;q10) in skeletal and
extraskeletal mesenchymal chondrosarcoma. Moderm Pathology 2002;
15: 572-6.
-
Park YK, Park HR, Kim YW, Chi SG, Unni KK. Lack
of Bcl10 mutations in malignant cartilaginous tumors. Intern J
Molecular Med 2002; 9:217-9.
-
Sakamoto A, Oda Y, Iwamoto Y, Tsuneyoshi M.
Expression of membrane type 1 matrix metalloprotease, matrix
metalloprotease 2 and tissue inhibitor of metalloprotease 2 in
human cartillaginous tumors with special emphasis on mesenchymal
and dedifferentiated chondrosarcoma. J Cancer Research &
Clinical Oncol 1999; 125: 541-8.
-
Szymanska J, Tarkkanen M, Wiklund T, Virolainen
M, Blomqvist C, Asko-Seljavaara S, Tukiainen E, Elomaa I,
Knuutila S. Cytogenetic study of extraskeletal mesenchymal
chondrosarcoma. A case report. Cancer Genetics &
Citogenetics. 1996; 86:170-3.
-
Castresana JS, Barrios C, Gomez L, Kreicbergs A.
Amplification of the c-myc proto-oncogene in human
chondrosarcoma. Diagnostic Molecular Pathology 1992; 1 :235-8.
-
Bruns J, Elbracht M, Niggemeyer O.
Chondrosarcoma of bone: an oncological and functional follow-up
study. Annals of Oncology 2001; 12:859-64.
-
Taconis WK, van der Heul RO,Taminiau AM.
Synovial chondrosarcoma: report of a case and review of the
literature. Skeletal Radiology 1997; 26: 682-5.
-
Anract P, Katabi M, Forest M, benoit J, Witvoet
J, Tomeno B. Synovial chondromatosis and chondrosarcoma. A study
of the relationship between these two diseases.Rev Chir Orthop
et Reparatrice de lAppareil Locomoteur 1996; 82:216-24.
-
Bertoni F, Unni KK, Beabout JW, Sim FH.
Chondrosarcomas of the synovium. Cancer 1991; 67: 155-62.
-
Laus M, Capanna R. Synovial chondromatosis and
chondrosarcoma of the hip: indications for surgical treatment.
Italian J Orthop & Traumatol 1982; 8:193-8.
-
Hermann G, Klein MJ, Abdelwahab IF, Kenan S.
Synovial chondrosarcoma arising in synovial chondromatosis of
the rigth hip. Skeletal Radiology 1997; 26: 366-9.
-
King JW, Spujut HJ, Fechner RE, Vanderpool DW.
Synovial chondrosarcoma of the knee joint. J Bone Joint Surg
1967; 49A: 1389-96.
-
Taras JS, Meadows SE. Synovial chondromatosis in
the radiocarpal joint.Am J Orthop 1995; 24:159-60.
-
Holm CL. Primary synovial chondromatosis of the
ankle. A case report. J Bone Joint Surg 1976; 58 B:878-80.
-
Wenger DE, Sundaram M, Unni KK, Janney CG,
Merkel K. Acral synovial chondrosarcoma. Skeletal Radiology
2002;. 31:125-9.
-
Cheung CC, Kandel AR, Bell RS, Mathews RE,
Ghazarian D. Extraskeletal Ewing sarcoma in a 77-year-old woman.
2001;125:
1358-61.
-
Falk S, Alpert M. Five-year survival of patients
with Ewing's sarcoma. Surg Gynecol Obstet 1967;124:319-324.
-
Bacci G, Ferrari S, Bertoni F, et al. Prognostic
factors in nonmetastatic Ewing's sarcoma of bone treated with
adjuvant chemotherapy. J Clin Oncol. 2000;18:4-11.
-
Hayes FA, Thompson El, Meyer WH. Therapy
for localized Ewing's sarcoma of bone. J Clin Oncol.
1989;7:208-213.
-
Nesbit ME Jr, Gehan EA, Burgert ED Jr.
Multimodal therapy for the management of primary, nonmetastatic
Ewing's sarcoma of bone: a long-term follow-up of the First
Intergroup study. JClin Oncol 1990;8:1664-1674.
-
Snkovics JG, Plager C, Ayala AG, Lindberg RD,
Samuels ML. Ewing sarcoma: its course and treatment in 50 adult
patients. Oncology 1980;37:114-119.
-
Siegel RD, Ryan LM, Antman KH. Adults with
Ewing's sarcoma: an analysis of 16 patients at the Dana-Farber
Cancer Institute. Am J Clin Oncol 1988;11:614-617.
-
Bacci G, Ferrari S, Comandone A. Neoadjuvant
chemotherapy for Ewing's sarcoma of bone in patients older than
thirty-nine years. Acta Oncol 2000;39:111-116.
-
Verrill MW, Judson IR, Wiltshaw E, Thomas A
Harmer CL, Fisher C. The use of paediatric chemotherapy
protocols at full dose is both a rational and feasible treatment
strategy in adults with Ewing's family tumours. Ann Oncol
1997;8:1099-1105.
|

